There is emerging evidence that cholinergic dysregulation (in particular, of
nicotinic cholinergic systems) may play a role in the pathophysiology of
attention-deficit/hyperactivity disorder (ADHD) [9]. Both clinical and epidemiological
experiments indicate that ADHD is associated with an increased risk and earlier age
of cigarette smoking [2, 7, 8]. A higher risk of smoking correlates directly with more
ADHD symptoms [2]. Furthermore, maternal smoking during pregnancy
increases the risk for ADHD in the offspring (independent of ADHD in mother)
[6].
2 Use of Nicotinic Receptor Agonists in Treatment of ADHD
Activating the cholinergic system can improve a host of cognitive processes (reviewed
by Levin et al. [4]) including learning, spatial and working memory, processing speed
and ability, inhibition, selective accuracy, detection, and overall attention. In general,
using the preclinical paradigms, the effects of nicotine appear to persist with chronic
administration [4]. Cholinergic pathways originating in the basal forebrain project
diffusely to the cerebral cortex [1]. acetylcholine (ACh) is particularly important for
normal cognitive function, including processes mediated by the prefrontal cortex that
are affected in ADHD, such as attention, working memory, and executive
function. Additionally, nicotinic acetylcholine receptor (nAChR) stimulation can
modulate dopaminergic neurotransmision [5, 10]. In animal experiments, nicotine
produces a concentration dependent release of dopamine (DA) from slices of
rat striatum [10], and nicotine has been shown to have effects on striatal
presynaptic dopamine transporters (DATs) in adults with ADHD [3]. However, not
all neuronal nAChR agonists reproduce the effects of nicotine on dopamine
release.
Longitudinal data continue to highlight the chronicity and clinical and public
health importance of ADHD and its treatment throughout the lifespan [11].
Unfortunately, many of the treatments for ADHD result in residual cognitive
symptoms. Hence, treatment strategies that include adequate treatment of
the general ADHD triad, and more specifically the attentional-based and
executive function symptoms of ADHD are needed [11]. Nicotinic cholinergic
neurotransmission plays an important role in attention and exectuve function
processes, and nicotine has demontrated procognitive effects in a number of
animal studies, and pilot data indicates some degree of efficacy in small
proof-of-concept adult ADHD trails [11]. Although adverse effects associated with
nicotine preclude its development as a therapeutic, a number of novel α4β2
nAChR agonists with improved safety/tolerability profiles have been discovered
[11].
Acronyms
ACh
acetylcholine
ADHD
attention-deficit/hyperactivity disorder
DAT
dopamine transporter
DA
dopamine
nAChR
nicotinic acetylcholine receptor
References
[1] J. L. Cummings and I. Litvan. Neuropsychiatric aspects of
corticobasal degeneration. Adv Neurol, 82:147–152, 2000.
[2] S. H. Kollins, F. J. McClernon, and B. F. Fuemmeler. Association
between smoking and attention-deficit/hyperactivity disorder symptoms in
a population-based sample of young adults. Arch. Gen. Psychiatry, 62(10):
1142–1147, Oct 2005.
[3] K. H. Krause, S. H. Dresel, J. Krause, H. F. Kung, K. Tatsch, and
M. Ackenheil. Stimulant-like action of nicotine on striatal dopamine
transporter in the brain of adults with attention deficit hyperactivity
disorder. Int. J. Neuropsychopharmacol., 5(2):111–113, Jun 2002.
[4] E. D. Levin, F. J. McClernon, and A. H. Rezvani. Nicotinic
effects on cognitive function: behavioral characterization, pharmacological
specification, and anatomic localization. Psychopharmacology (Berl.), 184
(3-4):523–539, Mar 2006.
[5] G. Mereu, K. W. Yoon, V. Boi, G. L. Gessa, L. Naes, and T. C.
Westfall. Preferential stimulation of ventral tegmental area dopaminergic
neurons by nicotine. Eur. J. Pharmacol., 141(3):395–399, Sep 1987.
[6] S. Milberger, J. Biederman, S. V. Faraone, L. Chen, and J. Jones.
Is maternal smoking during pregnancy a risk factor for attention deficit
hyperactivity disorder in children? Am J Psychiatry, 153(9):1138–42, Sep
1996.
[7] S. Milberger, J. Biederman, S. V. Faraone, L. Chen, and J. Jones.
ADHD is associated with early initiation of cigarette smoking in children
and adolescents. J Am Acad Child Adolesc Psychiatry, 36(1):37–44, Jan
1997.
[8] O. F. Pomerleau, K. K. Downey, F. W. Stelson, and C. S. Pomerleau.
Cigarette smoking in adult patients diagnosed with attention deficit
hyperactivity disorder. J Subst Abuse, 7(3):373–378, 1995.
[9] A. S. Potter, P. A. Newhouse, and D. J. Bucci. Central
nicotinic cholinergic systems: a role in the cognitive dysfunction in
attention-deficit/hyperactivity disorder? Behav. Brain Res., 175(2):
201–211, Dec 2006.
[10] T. C. Westfall, H. Grant, L. Naes, and M. Meldrum. The effect of
opioid drugs on the release of dopamine and 5-hydroxytryptamine from
rat striatum following activation of nicotinic-cholinergic receptors. Eur. J.Pharmacol., 92(1-2):35–42, Aug 1983.
[11] T. E. Wilens and M. W. Decker. Neuronal nicotinic receptor agonists
for the treatment of attention-deficit/hyperactivity disorder: focus on
cognition. Biochem. Pharmacol., 74(8):1212–1223, Oct 2007.
Age at the time of drug treatment and pharmacokinetic differences in absorption,
distribution and metabolism could influence both the acute and chronic effects of
psychostimulants [28]. For a 10 mg dose in a 30 kg child, the maximum serum
concentration occurs about 1.5 to 2 hours afterward and leads to concentrations of
approximately 10 ng/mL in plasma after administration, dropping by 50 % about 2
hours later [4]. However, the optimal therapeutic dose varies considerably across
individuals [9]. There is great variability among subjects and among dosing regimens
in the rate of absorption of methylphenidate (MPH) and the rate of decay of
MPH from plasma. This led to a 3.5-fold difference in blood levels between
subjects taking the same dose in the most extreme case [21]. The therapeutic
effects parallel the serum concentration of immediate-release MPH, with a
maximum reduction in attention-deficit/hyperactivity disorder (ADHD) symptoms
about 1.5 to 2 hours after dosing followed by a decline that is sufficient to
require another dose about 4 hours after the first to reestablish full efficacy
[23].
Oral MPH in normal adults has a half-life of about 2.1 h, the time to peak plasma
concentration is about 2.2 h. Ritalinic acid (major metabolite) reaches peak plasma
concentration at approximately the same time as MPH except that levels were
several-fold those of MPH [27]. Oral MPH in ADHD children isn’t too different: half-life
2.4 h; time to peak 1.5 h [27].
MPH undergoes extensive and stereospecific presystemic metabolism [1] to form
predominantly the inactive hydrolysis product, ritalinic acid, resulting in a low
absolute oral bioavailability. The bioavailability of oral MPH measured in rat is 0.19,
in monkey 0.22, with considerable intersubject variability: ranging from 0.08 to 0.44
in both species [27]. Animal studies have suggested that MPH is subject
to substantial first-pass and presystemic metabolism, the primary action
of presystemic metabolism is deesterification in the gut and/or intestinal
wall [1, 27]. Interestingly, the rate and extent of d-MPH absorption were
similar when administered with or without food [22]. MPH is metabolized
extensively in the rat; less than 1 % unchanged MPH was found in the 48 h urine
[7].
Intravenous administration of 1 mg/kg of MPH in rats shows rapid accumulation
of MPH in the brain, the peak brain concentration within the first minute after
injection [13]. Transfer of MPH from plasma to tissues is rapid and distribution is
extensive [7]. During the first 30 min after MPH administration, there was an average
of 8-fold greater MPH concentration in brain vs. plasma [13]. Peak serum and brain
concentrations after oral administration of 1 mg/kg of MPH occurred at 10 min after
dosing. At 45 min after dosing, the brain/serum ratios were approximately equal for
both i.v. and oral routes of administration [13]. The half-life of MPH in the rat
is 24.8 min [7]. MPH is ionized at physiological pH to a lesser extent than
dextroamphetamine (D-AMPH) and is considerably more lipid soluble, which will
induce quicker rise times to the brain [7].
After oral administration of [11C]d-threo-MPH, the radiolabeled d-MPH was
distributed stereoselectively in the rat brain, especially in the striatum. Further, it
was shown that the d-isomer binds specifically to dopamine (DA) uptake sites in the
striatum [2]. [11C]l-threo-MPH had an homogeneous distribution throughout
the brain (after i.v. administration) [26]. The rate of uptake in the brain
for the two enantiomers was eqivalent but the clearance was significantly
slower for [11C]d-threo-MPH than for the l-isomer. The therapeutic and
safety profile of d-MPH is similar to that of racemic d,l-MPH [15]. However,
l-MPH offers anxiolytic and antipsychotic activity, serves as an antidote for
stimulant overdose [3], reduces cocaine induced locomotor sensitization [6], and
pretreatment with l-MPH attenuates the motor activity response to d-MPH
[5].
The median effective dose (ED50) for dopamine transporter (DAT) blockade was
calculated to be 0.25 mg/kg oral MPH [24] and 0.075 mg/kg i.v. MPH [25]; the half
maximal inhibitory concentration (IC50) of MPH at DAT: 84 nm [8]. The ED50 estimate
for global norepinephrine transporter (NET) was 0.14 mg/kg ± 0.02 mg/kg MPH,
although the local ED50 did vary by brain region [10]; the IC50 of MPH at NET:
510 nm [8] If occupancy = [10], then the average efficacious maintenance
doses of MPH used in children and adults occupy 70 % to 80 % of NET but only 60 %
to 70 % of DAT, based on the estimated ED50 value of 0.14 mg/kg for NET and
0.25 mg/kg for DAT [24]. Interestingly, NET has greater affinity for DA than for
norepinephrine (NE) [12], and whether DAT or NET is the predominant protein
clearing DA depends on the abundance of the two transporters in a given region
[11].
1.1 Sustained-release preparations of MPH
In recent years, new drug delivery methods have been developed to enhance the
duration of action [21]. Through these efforts to develop effective long-acting MPH
preparations, valuable new information has become available on the relationship
between dosing regimens, blood levels, and therapeutic response. Efficacy of
MPH declined in an administration regimen designed to provide consistent
stable blood levels, presumably due to acute tolerance due to an adaptation
response at the synaptic level in response to blockade of the DAT [18, 20]. This
confirmed and extended findings of rapid tolerance previously observed by
Srinivas et al. [16] in an acute administration paradigm. This lead to the
discovery that there are two alterative strategies for providing sustained
efficacy: Pulsatile delivery or escalating blood levels [17]. With pulsatile
delivery, there is a drop in blood levels that enables the system to retain
or regain sensitivity. With escalating delivery, the increasing blood levels
presumably help to overcome emerging tolerance and waning sensitivity
[19].
1.2 Placebo effects of MPH
Subjects receiving placebo showed no significant degree of benefit and on average
were slightly worse following placebo administration than when they first arrived at
the laboratory in an unmedicated state [21]. ADHD does not appear to be a
placebo-responsive disorder when evaluated using objective measures. Instead, after
placebo administration, a clear pattern of deterioration across the day emerges,
in contrast to the pattern of improvement with MPH [18]. Improvement of
ADHD children on placebo in studies using observer ratings is probably due
in large part to a halo effect, in which teachers or parents score children
differently if they believe that they have been medicated [21]. Also, when MPH is
given therapeutically and produces large improvements in the classroom,
children with ADHD do not attribute this ‘success’ to the drug [14]. This
indicates that expectancy does not play a prominent role in MPH’s therapeutic
effects.
Acronyms
ADHD
attention-deficit/hyperactivity disorder
D-AMPH
dextroamphetamine
DAT
dopamine transporter
DA
dopamine
ED50
median effective dose
IC50
half maximal inhibitory concentration
MPH
methylphenidate
NET
norepinephrine transporter
NE
norepinephrine
References
[1] T. Aoyama, H. Kotaki, and T. Iga. Dose-dependent kinetics
of methylphenidate enantiomers after oral administration of racemic
methylphenidate to rats. J Pharmacobiodyn, 13(10):647–52, Oct 1990.
[2] T. Aoyama, H. Kotaki, Y. Sawada, and T. Iga. Stereospecific
distribution of methylphenidate enantiomers in rat brain: specific binding
to dopamine reuptake sites. Pharm Res, 11(3):407–11, Mar 1994.
[3] R. Baldessarini and A. Campbell. Method of dopamine inhibition
using l-threo-methylphenidate, Apr. 2001. US Patent 6,221,883.
[4] Y. P. Chan, J. M. Swanson, S. S. Soldin, J. J. Thiessen, S. M.
Macleod, and W. Logan. Methylphenidate hydrochloride given with or
before breakfast: Ii. effects on plasma concentration of methylphenidate
and ritalinic acid. Pediatrics, 72(1):56–9, Jul 1983.
[5] E. Davids, K. Zhang, N. S. Kula, F. I. Tarazi, and R. J. Baldessarini.
Effects of norepinephrine and serotonin transporter inhibitors on
hyperactivity induced by neonatal 6-hydroxydopamine lesioning in rats. JPharmacol Exp Ther, 301(3):1097–102, Jun 2002.
[6] Y.-S. Ding, S. J. Gatley, P. K. Thanos, C. Shea, V. Garza, Y. Xu,
P. Carter, P. King, D. Warner, N. B. Taintor, D. J. Park, B. Pyatt, J. S.
Fowler, and N. D. Volkow. Brain kinetics of methylphenidate (ritalin)
enantiomers after oral administration. Synapse, 53(3):168–75, Sep 2004.
doi: 10.1002/syn.20046.
[7] J. Gal, B. J. Hodshon, C. Pintauro, B. L. Flamm, and A. K. Cho.
Pharmacokinetics of methylphenidate in the rat using single-ion monitoring
glc-mass spectrometry. J Pharm Sci, 66(6):866–9, Jun 1977.
[8] S. J. Gatley, N. D. Volkow, A. N. Gifford, J. S. Fowler, S. L.
Dewey, Y. S. Ding, and J. Logan. Dopamine-transporter occupancy after
intravenous doses of cocaine and methylphenidate in mice and humans.
Psychopharmacology (Berl), 146(1):93–100, Sep 1999.
[9] L. L. Greenhill, J. M. Swanson, B. Vitiello, M. Davies, W. Clevenger,
M. Wu, L. E. Arnold, H. B. Abikoff, O. G. Bukstein, C. K. Conners,
G. R. Elliott, L. Hechtman, S. P. Hinshaw, B. Hoza, P. S. Jensen,
H. C. Kraemer, J. S. March, J. H. Newcorn, J. B. Severe, K. Wells,
and T. Wigal. Impairment and deportment responses to different
methylphenidate doses in children with adhd: the mta titration trial. JAm Acad Child Adolesc Psychiatry, 40(2):180–7, Feb 2001. doi: 10.1097/
00004583-_200102000-_00012.
[10] J. Hannestad, J.-D. Gallezot, B. Planeta-Wilson, S.-F. Lin, W. A.
Williams, C. H. van Dyck, R. T. Malison, R. E. Carson, and Y.-S.
Ding. Clinically relevant doses of methylphenidate significantly occupy
norepinephrine transporters in humans in vivo. Biol Psychiatry, 68(9):
854–60, Nov 2010. doi: 10.1016/j.biopsych.2010.06.017.
[11] J. A. Morón, A. Brockington, R. A. Wise, B. A. Rocha, and B. T.
Hope. Dopamine uptake through the norepinephrine transporter in
brain regions with low levels of the dopamine transporter: evidence from
knock-out mouse lines. J Neurosci, 22(2):389–95, Jan 2002.
[12] T. Pacholczyk, R. D. Blakely, and S. G. Amara. Expression cloning of
a cocaine- and antidepressant-sensitive human noradrenaline transporter.
Nature, 350(6316):350–4, Mar 1991. doi: 10.1038/350350a0.
[13] K. S. Patrick, K. R. Ellington, and G. R. Breese. Distribution of
methylphenidate and p-hydroxymethylphenidate in rats. J Pharmacol ExpTher, 231(1):61–5, Oct 1984.
[14] W. E. Pelham, B. Hoza, H. L. Kipp, E. M. Gnagy, and S. T.
Trane. Effects of methylphenidate and expectancy of ADHD children’s
performance, self-evaluations, persistence, and attributions on a cognitive
task. Exp Clin Psychopharmacology, 5:3–13, 1997.
[15] D. Quinn, S. Wigal, J. Swanson, S. Hirsch, Y. Ottolini, M. Dariani,
M. Roffman, J. Zeldis, and T. Cooper. Comparative pharmacodynamics
and plasma concentrations of d-threo-methylphenidate hydrochloride after
single doses
of d-threo-methylphenidate hydrochloride and d,l-threo-methylphenidate
hydrochloride in a double-blind, placebo-controlled, crossover laboratory
school study in children with attention-deficit/hyperactivity disorder. JAm Acad Child Adolesc Psychiatry, 43(11):1422–9, Nov 2004. doi: 10.1097/
01.chi.0000140455.96946.2b.
[16] N. R. Srinivas, J. W. Hubbard, D. Quinn, and
K. K. Midha. Enantioselective pharmacokinetics and pharmacodynamics
of dl-threo-methylphenidate in children with attention deficit hyperactivity
disorder. Clin Pharmacol Ther, 52(5):561–8, Nov 1992.
[17] J. Swanson, S. Gupta, A. Lam, I. Shoulson, M. Lerner, N. Modi,
E. Lindemulder, and S. Wigal.
Development of a new once-a-day formulation of methylphenidate for the
treatment of attention-deficit/hyperactivity disorder: proof-of-concept and
proof-of-product studies. Arch Gen Psychiatry, 60(2):204–11, Feb 2003.
[18] J. M. Swanson, S. Gupta, L. Williams, D. Agler, M. Lerner, and
S. Wigal. Efficacy of a new pattern of delivery of methylphenidate for the
treatment of ADHD: effects on activity level in the classroom and on the
playground. J Am Acad Child Adolesc Psychiatry, 41:1306–1314, 2002.
[19] J. M. Swanson, B. J. Casey, J. Nigg, F. X. Castellanos, N. D. Volkow,
and E. Taylor. Clinical and cognitive definitions of attention deficits in
children with attention-deficit/hyperactivity disorder. In M. I. Posner,
editor, Cognitive neuroscience of attention, pages 430– 445. Guilford, New
York, NY, 2004.
[20] J. L. Swanson-Park, C. M. Coussens, S. E. Mason-Parker, C. R.
Raymond, E. L. Hargreaves, M. Dragunow, A. S. Cohen, and W. C.
Abraham. A double dissociation within the hippocampus of dopamine
d1/d5 receptor and beta-adrenergic receptor contributions to the
persistence of long-term potentiation. Neuroscience, 92(2):485–97, 1999.
[21] M. H. Teicher, A. Polcari, M. Foley, E. Valente, C. E. McGreenery,
W.-W. Chang, G. McKay, and K. K. Midha. Methylphenidate blood levels
and therapeutic response in children with attention-deficit hyperactivity
disorder: I. effects of different dosing regimens. J Child AdolescPsychopharmacol, 16(4):416–31, Aug 2006. doi: 10.1089/cap.2006.16.416.
[22] S. K. Teo, M. R. Scheffler, A. Wu, D. I. Stirling, S. D. Thomas,
D. Stypinski, and V. D. Khetani. A single-dose, two-way crossover,
bioequivalence study of dexmethylphenidate hcl with and without food
in healthy subjects. J Clin Pharmacol, 44(2):173–8, Feb 2004. doi:
10.1177/0091270003261899.
[23] N. D. Volkow and J. M. Swanson. Variables that affect the clinical use
and abuse of methylphenidate in the treatment of ADHD. Am J Psychiatry,
160(11):1909–1918, Nov 2003.
[24] N. D. Volkow, G. J. Wang, J. S. Fowler, S. J. Gatley, J. Logan, Y. S.
Ding, R. Hitzemann, and N. Pappas. Dopamine transporter occupancies
in the human brain induced by therapeutic doses of oral methylphenidate.
Am J Psychiatry, 155(10):1325–1331, Oct 1998.
[25] N. D. Volkow, G. J. Wang, J. S. Fowler, M. Fischman, R. Foltin,
N. N. Abumrad, S. J. Gatley, J. Logan, C. Wong, A. Gifford, Y. S. Ding,
R. Hitzemann, and N. Pappas. Methylphenidate and cocaine have a
similar in vivo potency to block dopamine transporters in the human brain.
Life Sci, 65(1):PL7–12, 1999.
[26] N. D. Volkow, J. S. Fowler, G.-J. Wang, Y. S. Ding, and S. J.
Gatley. Role of dopamine in the therapeutic and reinforcing effects
of methylphenidate in humans: results from imaging studies. EuropeanNeuropsychopharmacology, 12:557–566, 2002.
[27] W. Wargin, K. Patrick, C. Kilts, C. T. Gualtieri, K. Ellington,
R. A. Mueller, G. Kraemer, and G. R. Breese. Pharmacokinetics of
methylphenidate in man, rat and monkey. J Pharmacol Exp Ther, 226(2):
382–6, Aug 1983.
[28] P. B. Yang, A. C. Swann, and N. Dafny. Dose-response characteristics
of methylphenidate on locomotor behavior and on sensory evoked potentials
recorded from the vta, nac, and pfc in freely behaving rats. Behav BrainFunct, 2:3, 2006. doi: 10.1186/1744-_9081-_2-_3.
The etiology of attention-deficit/hyperactivity disorder (ADHD) is unclear. However,
it is well accepted that ADHD has both genetic and environmental risk factors [3, 13].
ADHD is likely not a single pathophysiological entity, which suggests complex and
multiple etiologies. Multiple genetic and environmental factors may act in concert,
which gives rise to a spectrum of neurobiological liability [10] which manifests as a
set of ADHD symptoms. Estimates of heritability from many familial and twin studies
range from 0.5 to 0.9, average 0.75; which, taken together with a five-fold increased
risk in first-degree relatives, strongly implies a genetic component [3, 8, 10, 13, 30].
Environmental risk factors such as obstetric complications, family conflict,
and lead exposure have also shown positive, yet small, increases in risk for
ADHD.
2 Genetic Risk Factors
No single gene abnormality reliably predicts ADHD, but consistent association
between several genes regulating dopamine (DA) transport and signaling have been
implicated (these have been the genes most studied by candidate gene association
studies because of prevalent DA hypotheses concerning the pathophysiology of ADHD
[19, 37] and the mechanisms of the most popular drugs used to treat ADHD
[32, 34–36] ). Genome-wide association studies have failed to report any associations
that are significant after correction for multiple testing [10]. Cortese et al. [8]
estimates that a genome-wide association study would have to use sample sizes
between 10,000 to 20,000 subjects in order to accurately detect novel genes involved
in ADHD with risk factors similar to those seen in candidate gene association
studies. Candidate gene association studies have shown positive, but small
correlations with the dopamine receptor D4 gene (DRD4), dopamine receptor D5
gene (DRD5), dopamine transporter gene (SLC6A3), dopamine β-hydroxylase (DβH),
synaptosomal-associated protein of 25 kDa gene (SNAP25), serotonin transporter
gene (SLC6A4), and the serotonin 1B receptor gene (HTR1B) [3], which is in line
with the multifactorial polygenic model in which a plethora of genes each
confer a small but real risk to the disorder [8]. It is easy to imagine that
combinations of risk alleles impacting dopaminergic signaling could produce an
extreme hypodopaminergic state that would lead to poor cognitive function
[30].
The SLC6A3 and DRD4 genes show variation amongst the population based on a
variable number of tandem repeats where the nucleotide or base pair sequence is
repeated a different number of times in different alleles of the gene. A 40 base pair
variable tandem nucleotide repeat in the 3′ untranslated region of the human
DAT1 gene (SLC6A3 [7]) and a 48 base pair DA D4 receptor 7-repeat allele [17] have
both been implicated in ADHD, and have been the most consistently positive across
multiple candidate gene association studies [13, 14, 20, 29], which is not always
common [6, 9].
Dopamine receptor genes linked to ADHD
The DA D4 receptor is prevalently expressed in frontal and subcortical
networks that are implicated in the pathophysiology of ADHD [3]. DRD4
association studies have assayed a variant known as the exon-III 7-repeat allele
(16 amino acid repeat in the 3rd intracellular loop of the receptor that
couples the receptor to pre- or post-synaptic G-protein effectors [30]), which
while it shows a positive association with ADHD, may paradoxically be
protective, as the DRD4 7-repeat allele produces an in vitro blunted response to
dopamine, but subjects with the DRD4 7-repeat allele and ADHD tend to
have better clinical and academic outcomes than those with ADHD and the
DRD4 4-repeat allele more common in the general population [3, 17, 30].
Candidate gene studies of the DA D5 receptor, which is expressed in the
hippocampus, and especially the dentate gyrus [16], have shown positive
correlation with ADHD with a repeated sequence near the transcription start site
[3].
Although DA D1 and D2 receptors are thought to be the major effectors of
stimulant-induced alterations in impulsive, addictive, and compulsive behavior [33], it
is unsettled as to whether variants of these full-length genes are associated with
ADHD [27].
Dopamine transporter gene linked to ADHD
When pooled in meta-analysis, studies of the SLC6A3 10-repeat sequence in the
3′ untranslated region (the 3′ untranslated region of mRNA often contains
regulatory sequences that can influence post-transcriptional gene expression)
estimate an increased odds ratio of 1.13 for ADHD [3, 7]. It may be possible
that different variants of the dopamine transporter (DAT) have an altered
sensitivity to endogenous DA. At present, no such variations in the coding
sequence of the DAT are known, nor are they separable pharmacologically
[27].
Norepinephrine synthesis linked to ADHD
Dopamine β-hydroxylase is the main enzyme and rate-limiting step in converting
DA to norepinephrine (NE). Meta-analysis of multiple family studies suggest a
significant association with ADHD and the 5′ Taq1 polymorphism of the DβH gene
[3].
Serotonin signaling linked to ADHD
Both the serotonin transporter gene (SLC6A4) and the serotonin 1B receptor
gene (HTR1B) have been implicated in ADHD. A functional variant of the SLC6A4 has
a meta-analysis adjusted odds ratio of 1.31 for ADHD. Many studies of the HTR1B
have also been positive, with a non-functional marker showing an odds ratio of 1.44
[3].
Synaptic transmission linked to ADHDSNAP25 is a neuron-specific protein involved in synaptic vesicle transport, fusion,
and release. A meta-analysis of several studies assessing the association between the
gene and ADHD found an odds ratio of 1.19. SNAP25 knockout mice also
show spontaneous hyperactivity which can be reduced with stimulant drugs
[3].
Negative candidate gene results
Candidate gene association studies have also been done on several genes
that have provided negative or equivocal results. Two genes involved in
monoamine degredation, catechol-O-methyl-transferase (COMT) and monoamine
oxidase (MAO), have shown negative results, as well as the norepinephrine
transporter gene (SLC6A2) and the norepinephrine 1C, 2A, and 2C receptors
[3].
3 Environmental Risk Factors
Environmental factors play a critical role in the etiology of ADHD. One of the
best evidences supporting this point comes from the genetic twin studies,
which show that the risk of ADHD in monozygotic twins is much lower than
100 % [8]. Many studies have indicated that complications during pregnancy
and delivery could be associated with childhood ADHD. The complications
that are associated with later ADHD tend to include chronic exposures to
the fetus, and complications that lead to hypoxia rather than acute events
[3].
Prenatal risk factors
Maternal alcohol use during pregnancy leads to behavioral, cognitive, and
learning problems that could present as ADHD. Prenatal alcohol exposure is known
to induce brain structural anomalies, and children exposed to prenatal alcohol are
more hyperactive, disruptive, impulsive, and are at increased risk of a range of
psychiatric disorders [10]. Studies of ADHD children show an increased likelihood of
having been exposed to alcohol as a fetus [3].
There is also a well documented, almost 3-fold increased risk for the disorder in
children whose mothers smoked during pregnancy [3, 24]. Exposure of the fetus to
nicotine can damage the development of the brain at critical time points as nicotinic
receptors modulate dopaminergic activity [11, 12, 31], and animal studies have also
shown a correlation between chronic nicotine exposure of pregnant dams and
hyperactive offspring [3].
Perinatal risk factors
Low birthweight (< 2500g) and premature birth (< 37weeks gestation) increase
the risk for ADHD [2, 3, 18, 22]. Very low birthweight children show a 2-fold increase
in the proportion of those with ADHD vs. average birthweight counterparts [10].
Interestingly, prenatal tobacco exposure can result in both low birthweight and
preterm birth [15]. In a huge study from the Danish longitudinal birth registers,
Linnet et al. [21] found a rate ratio for hyperkinetic disorder (the International
Statistical Classification of Diseases and Related Health Problems (ICD)
analogue of the Diagnostic and Statistical Manual of Mental Disorders, Fourth
Edition (DSM-IV)’s ADHD) of 1.7 for 34 to 37 weeks gestation and rate ratio 2.7 for
less than 34 weeks gestation. Likewise, low birth weight for children born at
term gave a rate ratio of 1.9 for birthweight of 1500 g to 2499 g and 1.5 for
birthweight 2500 g to 2999 g [21]. In a similar study, Mick et al. [23] reported
that ADHD cases were 3.1 times more likely than controls to be born under
2500 g.
Postnatal and childhood risk factors
Malnutrition and dietary deficiency have been proposed, and iron deficiency has
been implicated in some cases of ADHD, however further evidence is necessary to
firmly establish a role [10].
Lead exposure
Lead exposure has been implicated in the pathophysiology of ADHD, although
most children with ADHD do not show lead contamination and many children
with high lead exposure do not become affected with the disorder [3]. Nigg
et al. [26] suggests that low-level lead exposure, below the US Environmental
Protection Agency (EPA) limits, may represent a hidden major effect on ADHD
incidence. A population sample, Braun et al. [4] reported that very low
levels of lead were associated with ADHD, even with lead exposures below
5 μg/dL, which highlights the importance of a gene–environment interaction
model, as these levels of lead exposure are common in the US population
[5].
4 Gene–Environment Interactions
Molecular genetics supports the association of ADHD with several DA signaling
related genes, and environmental effects suggests increased risks for ADHD related to
maternal smoking, exposure to low levels of lead, premature birth or low birth
weight, and other factors that alter fetal development with lasting or possibly
permanent effects on attention and behavior [30]. It is noteworthy that individual
genes or environmental risk factors carry quite modest risk, and also that some of the
environmental risk factors associated with ADHD (e.g. low lead exposure) are quite
common in the general population [30]. More complex models of the etiology of
ADHD which incorporate gene–environment interactions are being studied. However,
the multitude of possible etiologies of gene–environment combinations add
complexity, and would require enormous sample sizes for adequate evaluation [30].
Neuman et al. [25] demonstrated that smoking during pregnancy is associated with
the ADHD combined subtype in children with the ‘susceptible’ DRD4 and dopamine
transporter gene (SLC6A3) gene variants. Also, a significant interaction between
SLC6A3 genotype and prenatal smoke exposure was found in males: Men with
prenatal smoke exposure and homozygous for the SLC6A3 10-repeat allele had higher
hyperactivity/impulsivity than males from all other groups [1]. Through unknown
mechanisms, abnormal DAT density is a common feature among subjects with ADHD
[28].
Acronyms
ADHD
attention-deficit/hyperactivity disorder
CDC
Center for Disease Control and Prevention
COMT
catechol-O-methyl-transferase
DAT
dopamine transporter
DA
dopamine
DbH
dopamine β-hydroxylase
DRD4
dopamine receptor D4 gene
DRD5
dopamine receptor D5 gene
DSM-IV
Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition
EPA
US Environmental Protection Agency
HTR1B
serotonin 1B receptor gene
ICD
International Statistical Classification of Diseases and Related Health
Problems
MAO
monoamine oxidase
NE
norepinephrine
SLC6A2
norepinephrine transporter gene
SLC6A3
dopamine transporter gene
SLC6A4
serotonin transporter gene
SNAP25
synaptosomal-associated protein of 25 kDa gene
References
[1] K. Becker, M. El-Faddagh, M. H. Schmidt, G. Esser, and M. Laucht.
Interaction of dopamine transporter genotype with prenatal smoke
exposure on adhd symptoms. J Pediatr, 152(2):263–9, Feb 2008. doi:
10.1016/j.jpeds.2007.07.004.
[2] A. T. Bhutta, M. A. Cleves, P. H. Casey, M. M. Cradock, and
K. J. S. Anand. Cognitive and behavioral outcomes of school-aged children
who were born preterm: a meta-analysis. JAMA, 288(6):728–37, Aug 2002.
[4] J. M. Braun, R. S. Kahn, T. Froehlich, P. Auinger, and B. P.
Lanphear. Exposures to environmental toxicants and attention deficit
hyperactivity disorder in u.s. children. Environ Health Perspect, 114(12):
1904–9, Dec 2006.
[5] Center for Disease Control and Prevention (CDC). Blood lead
levels–united states, 1999-2002. MMWR. Morbidity and mortality weeklyreport, 54(20):513, 2005.
[6] D. A. Collier, S. Curran, and P. Asherson. Mission: not impossible?
candidate gene studies in child psychiatric disorders. Mol Psychiatry, 5(5):
457–60, Sep 2000.
[7] E. H. Cook, Jr, M. A. Stein, M. D. Krasowski, N. J. Cox, D. M.
Olkon, J. E. Kieffer, and B. L. Leventhal. Association of attention-deficit
disorder and the dopamine transporter gene. Am J Hum Genet, 56(4):
993–8, Apr 1995.
[8] S. Cortese, S. V. Faraone, and J. Sergeant. Misunderstandings of the
genetics and neurobiology of adhd: moving beyond anachronisms. AmJ Med Genet B Neuropsychiatr Genet, 156B(5):513–6, Jul 2011. doi:
10.1002/ajmg.b.31207.
[9] R. R. Crowe. Candidate genes in psychiatry: an epidemiological
perspective. Am J Med Genet, 48(2):74–7, Jul 1993. doi: 10.1002/ajmg.
1320480203.
[10] P. Curatolo, E. D’Agati, and R. Moavero. The neurobiological basis
of adhd. Ital J Pediatr, 36(1):79, 2010. doi: 10.1186/1824-_7288-_36-_79.
[11] J. A. Dani, D. Ji, and F.-M. Zhou. Synaptic plasticity and nicotine
addiction. Neuron, 31(3):349–352, 2001.
[12] J. A. Dani, D. Jenson, J. I. Broussard, and
M. De Biasi. Neurophysiology of nicotine addiction. Journal of addictionresearch & therapy, 1(1), 2011.
[13] S. DiMaio, N. Grizenko, and R. Joober. Dopamine genes and
attention-deficit hyperactivity disorder: a review. J Psychiatry Neurosci,
28(1):27–38, Jan 2003.
[14] S. V. Faraone, A. E. Doyle, E. Mick, and J. Biederman. Meta-analysis
of the association between the 7-repeat allele of the dopamine d(4) receptor
gene and attention deficit hyperactivity disorder. Am J Psychiatry, 158(7):
1052–7, Jul 2001.
[15] J. J. Jaakkola, N. Jaakkola, and K. Zahlsen. Fetal growth and length
of gestation in relation to prenatal exposure to environmental tobacco
smoke assessed by hair nicotine concentration. Environ Health Perspect,
109(6):557–61, Jun 2001.
[16] Z. U. Khan, L. Mrzljak, A. Gutierrez, A. de la Calle, and P. S.
Goldman-Rakic. Prominence of the dopamine d2 short isoform in
dopaminergic pathways. Proc Natl Acad Sci U S A, 95(13):7731–6, Jun
1998.
[17] G. J. LaHoste, J. M. Swanson, S. B. Wigal, C. Glabe, T. Wigal,
N. King, and J. L. Kennedy. Dopamine d4 receptor gene polymorphism is
associated with attention deficit hyperactivity disorder. Mol Psychiatry, 1
(2):121–4, May 1996.
[18] J. Lahti, K. Räikkönen, E. Kajantie, K. Heinonen, A.-K. Pesonen,
A.-L. Järvenpää, and T. Strandberg. Small body size at birth and
behavioural symptoms of adhd in children aged five to six years. J ChildPsychol Psychiatry, 47(11):1167–74, Nov 2006. doi: 10.1111/j.1469-_7610.
2006.01661.x.
[19] F. Levy. The dopamine theory of attention deficit hyperactivity
disorder (adhd). Aust N Z J Psychiatry, 25(2):277–83, Jun 1991.
[20] J. Li, Y. Wang, R. Zhou, H. Zhang, L. Yang, B. Wang, and S. V.
Faraone. Association between polymorphisms in serotonin 2c receptor
gene and attention-deficit/hyperactivity disorder in han chinese subjects.
Neurosci Lett, 407(2):107–11, Oct 2006. doi: 10.1016/j.neulet.2006.08.022.
[21] K. M. Linnet, K. Wisborg, C. Obel, N. J. Secher, P. H. Thomsen,
E. Agerbo, and T. B. Henriksen. Smoking during pregnancy and the risk
for hyperkinetic disorder in offspring. Pediatrics, 116(2):462–7, Aug 2005.
doi: 10.1542/peds.2004-_2054.
[22] K. M. Linnet, K. Wisborg, E. Agerbo, N. J. Secher, P. H. Thomsen,
and T. B. Henriksen. Gestational age, birth weight, and the risk of
hyperkinetic disorder. Arch Dis Child, 91(8):655–60, Aug 2006. doi:
10.1136/adc.2005.088872.
[23] E. Mick, J. Biederman, J. Prince, M. J. Fischer, and S. V. Faraone.
Impact of low birth weight on attention-deficit hyperactivity disorder. JDev Behav Pediatr, 23(1):16–22, Feb 2002.
[24] S. Milberger, J. Biederman, S. V. Faraone, L. Chen, and J. Jones.
Is maternal smoking during pregnancy a risk factor for attention deficit
hyperactivity disorder in children? Am J Psychiatry, 153(9):1138–42, Sep
1996.
[25] R. J. Neuman, E. Lobos, W. Reich, C. A. Henderson, L.-W. Sun,
and R. D. Todd. Prenatal smoking exposure and dopaminergic genotypes
interact to cause a severe adhd subtype. Biol Psychiatry, 61(12):1320–8,
Jun 2007. doi: 10.1016/j.biopsych.2006.08.049.
[26] J. T. Nigg, M. M. Wong, M. M. Martel, J. M. Jester, L. I. Puttler,
J. M. Glass, K. M. Adams, H. E. Fitzgerald, and R. A. Zucker. Poor
response inhibition as a predictor of problem drinking and illicit drug use
in adolescents at risk for alcoholism and other substance use disorders.
J Am Acad Child Adolesc Psychiatry, 45(4):468–75, Apr 2006. doi:
10.1097/01.chi.0000199028.76452.a9.
[27] P. Seeman and B. K. Madras. Anti-hyperactivity medication:
methylphenidate and amphetamine. Mol Psychiatry, 3:386–396, 1998.
[28] T. J. Spencer, J. Biederman, B. K. Madras, S. V. Faraone, D. D.
Dougherty, A. A. Bonab, and A. J. Fischman. In vivo neuroreceptor
imaging in attention-deficit/hyperactivity disorder: a focus on the
dopamine transporter. Biol Psychiatry, 57(11):1293–300, Jun 2005. doi:
10.1016/j.biopsych.2005.03.036.
[29] J. M. Swanson, P. Flodman, J. Kennedy, M. A. Spence, R. Moyzis,
S. Schuck, M. Murias, J. Moriarity, C. Barr, M. Smith, and M. Posner.
Dopamine genes and adhd. Neurosci Biobehav Rev, 24(1):21–5, Jan 2000.
[30] J. M. Swanson, M. Kinsbourne, J. Nigg, B. Lanphear, G. A.
Stefanatos, N. Volkow, E. Taylor, B. J. Casey, F. X. Castellanos, and
P. D. Wadhwa. Etiologic subtypes of attention-deficit/hyperactivity
disorder: brain imaging, molecular genetic and environmental factors and
the dopamine hypothesis. Neuropsychol Rev, 17(1):39–59, Mar 2007. doi:
10.1007/s11065-_007-_9019-_9.
[31] J. Tang and J. A. Dani. Dopamine enables in vivo synaptic
plasticity associated with the addictive drug nicotine. Neuron, 63
(5):673–682, Sep 2009. doi: 10.1016/j.neuron.2009.07.025. URL
http://dx.doi.org/10.1016/j.neuron.2009.07.025.
[32] N. D. Volkow, Y. S. Ding, J. S. Fowler, G. J. Wang, J. Logan,
J. S. Gatley, S. Dewey, C. Ashby, J. Liebermann, and R. Hitzemann.
Is methylphenidate like cocaine? Studies on their pharmacokinetics and
distribution in the human brain. Arch. Gen. Psychiatry, 52(6):456–463,
Jun 1995.
[33] N. D. Volkow, G. J. Wang, M. W. Fischman, R. W. Foltin, J. S.
Fowler, N. N. Abumrad, S. Vitkun, J. Logan, S. J. Gatley, N. Pappas,
R. Hitzemann, and C. E. Shea. Relationship between subjective effects of
cocaine and dopamine transporter occupancy. Nature, 386(6627):827–30,
Apr 1997. doi: 10.1038/386827a0.
[34] N. D. Volkow, G. J. Wang, J. S. Fowler, S. J. Gatley, J. Logan, Y. S.
Ding, R. Hitzemann, and N. Pappas. Dopamine transporter occupancies
in the human brain induced by therapeutic doses of oral methylphenidate.
Am J Psychiatry, 155(10):1325–1331, Oct 1998.
[35] N. D. Volkow, G. Wang, J. S. Fowler,
J. Logan, M. Gerasimov, L. Maynard, Y. Ding, S. J. Gatley, A. Gifford,
and D. Franceschi. Therapeutic doses of oral methylphenidate significantly
increase extracellular dopamine in the human brain. J Neurosci, 21(2):
RC121, Jan 2001.
[36] N. D. Volkow, J. S. Fowler, G.-J. Wang, and R. Z. Goldstein. Role of
dopamine, the frontal cortex and memory circuits in drug addiction: insight
from imaging studies. Neurobiol Learn Mem, 78(3):610–24, Nov 2002.
[37] P. H. Wender, R. S. Epstein, I. J. Kopin, and E. K. Gordon. Urinary
monoamine metabolites in children with minimal brain dysfunction. Am JPsychiatry, 127(10):1411–5, Apr 1971.
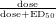 [
[